March 13, 2008
Study links cell death and inflammation
Cells are coded with several programs for self-destruction. Many cells die peacefully. Others cause a ruckus on their way out.
Some programmed cell death pathways simply and quietly remove unwanted cells, noted a team of UW researchers who study the mechanisms of cell destruction.
Then there is the screaming, alarm-ringing death of a potentially dangerous cell, such as a cell infected with Salmonella, they added. These dying cells spill chemical signals and get a protective response. The resulting inflammation, which the body launches in self-defense, can at times backfire and damage vital tissues.
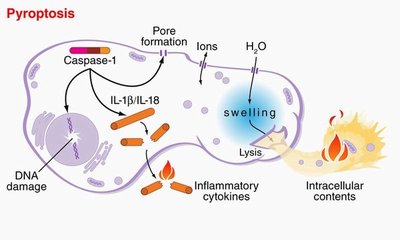
A research team lead by Dr. Brad T. Cookson, an associate professor of microbiology and laboratory medicine, named this type of cell death “pyroptosis,” Greek for going down in flames. Cell death that doesn’t cause inflammation is called “apoptosis”: to drop gently like leaves from a tree.
An enzyme inside cells, called caspase-1, plays a critical role in both harmful inflammation and in resistance to infection, Cookson and his colleagues noted. It’s not just responsible for cell death, but also for the production of inflammatory proteins that are released from the dying cell. Mice deficient in caspase-1 are susceptible to infection, yet resistant to toxic shock, tissue injury from lack of oxygen, and inflammatory bowel disease.
The Cookson lab has done many studies of caspase-1 and how it mediates the pathway of pro-inflammatory programmed cell death. The lab’s most recent study will be published the week of March 10 to March 14 in the online Early Edition of the Proceedings of the National Academy of Sciences. The study looked at how two different noxious stimuli, anthrax toxin and Salmonella infection, trigger the caspase-1-mediated cell death pathway. UW graduate students Susan Fink and Tessa Bergsbaken conducted this study.
The researchers found that each of these stimuli took an independent route to activate caspase-1; however, these two distinct mechanisms of activation eventually converged on a common pathway of cell death. This common pathway featured cleavage of the cell’s DNA, activation of inflammatory chemical messengers, and the final jettison of the cells contents. The spillage occurs after nano-scale pores form in the cell membrane, much like punctures in a water balloon.
According to Cookson, these findings are helping to create research models for studying a broadly important pathway of pro-inflammatory programmed cell death. The findings also support the notion that diverse disease agents can use different mechanisms to elicit this pathway.
“Examining this system provides insight into mechanisms of both beneficial and pathological cell death, and the strategies that infectious disease agents employ to manipulate the body’s responses,” Cookson said. His group’s previous studies of Yersinia, the plague pathogen, revealed that cell death mechanisms can be re-directed from a passive, non-inflammatory pathway, to a more beneficial inflammatory pathway. This finding suggests the possibility of treating diseases by modulating cell death pathways.
“In addition to its protective role in fighting infection,” Cookson added, “caspase-1 also plays a role in many medical conditions characterized by cell death and inflammation.” These conditions include organ damage in the heart, brain, lungs, nerves, and kidneys. Understanding pro-inflammatory cell death pathways may lead to new therapies against fatal or disabling diseases, such as serious infections, heart attack, cancer and stroke.
Cookson is part of the National Institutes of Health-funded Microscale Life Sciences Center, a collaboration among scientists and engineers from the UW, the University of Arizona, the Fred Hutchinson Cancer Research Center, and Brandeis University. The scientists work to discover basic mechanisms in the formation, growth, and decline of human cells. Their aim is to develop biotechnology to combat widespread diseases and environmental threats to human health.
