January 7, 2025
For the Record- January 7, 2025: DCT Guidance, Revised Short Form Policy, More
In this Issue:
Finalized Diversity in Clinical Trials Guidance and Plan
Effective January 1, 2026
In 2023 the WA State legislature passed 2SHB 1745, also known as the Diversity in Clinical Trials (DCT) bill. This bill aims to improve participation in clinical trials from underrepresented communities so that their data informs and contributes towards better health outcomes in these populations.
To comply with the requirements of the bill, HSD has developed a Diversity Plan for Clinical Trials. This form is intended to be used with the Diversity in Clinical Trials Guidance and includes links to relevant information.
For more details about the development of the guidance, refer to this background information and summary of key comments that shaped the guidance.
Key Information:
- UW Policy requires a Diversity Plan to improve the enrollment of underrepresented groups within the target study population for all research that meets the definition of a clinical trial and where UW employees or agents are responsible for or engaged in recruitment or consent activities.
- This policy applies:
- Regardless of where the interventions occur;
- To UW site(s) being reviewed by an external (non-UW) IRB; and
- To all sites as a condition of the UW IRB agreeing to review on behalf of non-UW institutions and individuals.
- In addition, the Diversity Plan includes new requirements for enrolling participants with a Non-English Language Preference (NELP).
What does this mean for studies reviewed by the UW IRB?
Applicable studies must submit the Diversity Plan as part of the Zipline application. When UW is the reviewing IRB for a multi-site trial, the requirement for all sites to comply with the policy will be a condition of the agreement for UW to serve as the reviewing IRB.
What does this mean for studies Reviewed by an External (non-UW) IRB?
The requirements differ depending on which external IRB is conducting the review.
- Local (WA State) Partners with Established Cooperative Agreements.
- When the review is conducted by Fred Hutchinson Cancer Center, Seattle Children’s, and Washington State IRBs, UW researchers do not need to complete the Diversity Plan as part of the Zipline request to use any of these external IRBs for review. The UW will defer to the policies developed by these cooperative partners to comply with the DCT bill.
- Commercial IRBs and Other External IRBs.
- When the review is conducted by WCG IRB or Advarra. UW researchers must include the Diversity Plan as part of the UW external review Zipline application and provide the supplement to WCG IRB or Advarra as part of their submission.
- When the review is conducted by an external IRB that is not WCG IRB, Advarra, or a cooperative partner listed above, UW researchers must include the Diversity Plan as part of the Zipline request to use an external (non-UW) IRB for review. The UW Reliance Team will assess the Diversity Plan for compliance with UW policy. Researchers then incorporate the Plan information into the external IRB’s application materials.
- For multi-site trials reviewed by commercial IRBs or other external IRBs, the diversity plan and policy requirements technically only apply to the UW site. However, in most cases it will make sense to discuss the target study population and enrollment goals in the context of the larger study and across sites.
One-year Transition Period. The effective date for compliance with the new Diversity in Clinical Trials requirements is January 1, 2026. The requirements will apply to new studies submitted to the IRB on or after the effective date. HSD acknowledges that researchers will need time to obtain the funding and resources to support these new requirements and we have planned for an effective date of one year from the publication of the Diversity Plan and associated guidance to allow for the preparatory work that will need to occur at pre-award/study design stage.
Resources. The UW Diversity in Clinical Trials Initiative has been working to develop pre- and post-award resources for researchers and the infrastructure to support the engagement of community partners. The Diversity in Clinical Trials guidance includes a section on Planning, Budgeting, and Resources which points to a number of these resources, some of which are still in development. HSD will provide additional information and updates as resources become available in the coming months.
Questions. Contact hsdinfo@uw.edu with any questions.
Revised Short Form Consent Policy Implemented
Reminder
Changes to HSD’s short form consent policy went into effect on January 1, 2025. These policy changes were announced in our November 5, 2024 Newsletter.
Short form consent is an alternative method of consent that is intended to be used only for the infrequent and unanticipated enrollment of an individual with a non-English language preference when there is not time and opportunity to obtain an appropriate written translation of the IRB-approved English consent form. This method involves presentation of the English consent form through a qualified interpreter and requires an observer to the consent process.
Key Changes.
- The new policy applies regardless of when the study was approved.
- Prospective IRB approval to use the short form method is now required for all greater than minimal risk studies and FDA-regulated studies reviewed by the UW IRB. In addition:
- Researchers must notify the IRB after use of the short form consent process and provide a translated consent form as a modification in Zipline within 30 days.
- Study participants must be provided with an IRB-approved translated consent form within 2 weeks of IRB approval. The translated consent form does not need to be signed and does not need to be provided to participants in person. It can, for example, be sent by email.
- In most instances, the required impartial witness must be proficient in the language of the oral consent presentation. However, there is some flexibility in certain circumstances when the interpreter is unable or unwilling to serve as the witness and there is no other impartial adult with sufficient language proficiency available.
- With the above additional requirements, HSD will no longer limit use of short form consent in greater than minimal risk research to only studies that provide the prospect of direct benefit that is not available outside the research.
What does this mean for ongoing research reviewed by a UW IRB?
- Ongoing studies that are greater than minimal risk or FDA regulated and plan to use the short form consent process must submit a study modification to obtain prospective IRB approval before using the short form consent process.
- Ongoing studies that involve minimal risk and are not FDA regulated do not need to obtain IRB approval to use the short form consent process. These studies are also not required to report use of this consent method to the IRB or provide participants with a translated consent form after its use, though provision of a translated consent form is encouraged.
What does this mean for research reviewed by an external (non-UW IRB)?
- UW researchers must follow the policies of the non-UW IRB regarding short forms, which may or may not include prospective IRB approval. HSD does not require prospective approval or a specific time frame for notification of the IRB when not required by the non-UW IRB.
- In addition to adhering to the non-UW IRB’s policy, when agents or employees of UW will be obtaining consent using the short form consent method for studies that are greater than minimal risk or FDA regulated:
- Researchers must submit a translated consent form to the reviewing IRB for approval within 30 days of using the short form consent process.
- The IRB-approved translated consent form must be provided to the participant within 2 weeks of IRB approval. UW does not specify the method by which the form is provided (e.g., email vs. paper) and does not require that the form be signed.
Questions. Please contact hsdinfo@uw.edu if you have questions.
Research Medicaid Attestation E-Signature Workflow
New
Starting January 13, 2025, a new E-Signature workflow for Research Medicaid Attestation forms will be required for UW and Fred Hutch researchers to complete in Epic. This only applies to study participants with Medicaid coverage being enrolled in a qualifying clinical trial (QCT). This new workflow utilizes electronic forms, automated In Basket routing, and electronic signatures in the collection of Medicaid Attestation forms.
This workflow is being implemented to streamline efforts related to research billing compliance requirements outlined in the UW Medicine COMP.202 policy. This policy was recently updated to reflect new federal requirements that the principal investigator (PI) must attest to the appropriateness of the QCT in which the individual is participating to the participant’s Medicaid plan (e.g., WA State Health Care Authority for Apple Health, or a Medicaid Managed Care plan, or an out-of-state Medicaid plan, etc.). Additionally, in situations where the PI is not also a study participant’s healthcare provider (HCP), both the PI and the HCP must sign the form.
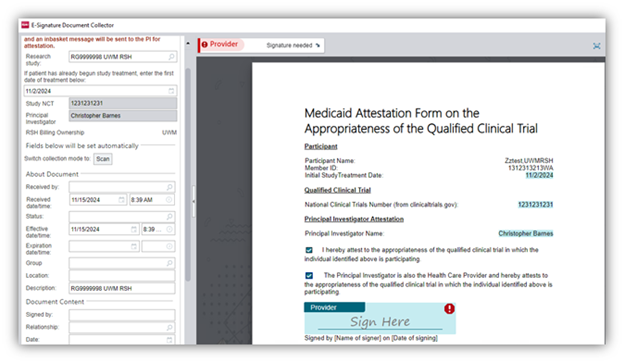
For additional project context, information, or training materials please refer to the UW Medicine/ Fred Hutch comprehensive training guide available on the EHR Hub (AMC log-in required).
Need Help?
If you have any workflow functionality questions or need further assistance, please call (206) 520-2200 to create a ticket and request the Epic Research team.
Compliance related questions should be directed to:
- Fred Hutch Compliance: integrity@fredhutch.org
- UW Medicine Compliance: comply@uw.edu | dorteroj@uw.edu
Certificate of Confidentiality & Third Parties
Update
A Certificate of Confidentiality (CoC) is a legal protection that some federal agencies can issue to researchers to protect identifiable sensitive information collected as part of a study.
Investigators and institutions have several responsibilities associated with the CoC. The NIH CoC website was recently updated to remind investigators of their responsibilities when using third party platforms/vendors:
Uphold CoC Protections: Institutions issued a CoC need to consider these protections when selecting third parties or entities (such as contractors and online platform vendors) and utilize those that can and will protect covered information against compelled disclosure. Remember that the institution holding the CoC is ultimately responsible for safeguarding all the information covered by the Certificate against compelled disclosure. For NIH funded studies, failure to comply with CoC protections is a violation of the terms and conditions of their award.
Terms that address the above requirement should be included in any contract and/or data processing/use agreement entered into with third parties.
We have updated the researcher responsibilities section of our CoC web guidance to reflect this requirement. Please contact us at hsdinfo@uw.edu if you have any questions.
Declaration of Helsinki
Revised
The World Medical Association (WMA) recently adopted revisions to the Declaration of Helsinki. The document was initially developed in 1964 as a statement of ethical principles for the conduct of medical research with human participants. Although not a legally binding document, it has greatly influenced U.S. and other international laws/regulations governing research. The latest version is available on the WMA website. For a discussion of some of the changes, check out this JAMA article.